Genome analysis strategies: a closer look at layer III of the genomic space
In this follow-up to our practical overview of the genomic space, we take a closer look at layer III and the considerations that impel its genomic analysis strategies.


To help stakeholders grasp a good understanding of the offerings in the genomics space so that the flurry of activity does not lead to confusion and stoppage of progress, we previously synthesized a practical overview of the genomic space. In this follow-up post, we will examine layer III of that overview a little more closely as it is in this layer that the quintessential problem of our time, the cipher of the genome function, resides. Given its inherent challenge and promise for great value creation for society, this layer is the most fraught with nuances and confusion.
Before going any further, let's define genetic feature, a term that we'll use a lot in this post. To aid comprehension, let's begin with genes, which everyone has heard of. Genes are segments of the genome that code for proteins and functional RNA. They make up about 1% of the genome and are themselves governed, controlled, and affected by a slew of other genetic entities (regulators, enhancers, repressors, etc.). When studying the exome alone, it may be sufficient to speak only of genes. However, when considering the whole genome in its entirety as we'll do here, it is more appropriate and complete to also consider the other elements found in the unexplored 99% of the genome that also affect gene expression. It is in this respect that we speak of genetic features to capture/denote all of the genomic entities (including genes) that affect the expression of an organism's phenotypes. A further level of depth is possible to distinguish between specific characteristic changes at these entities including copy number variation, expansions, repeats, insertions, deletions, and multi- and single base polymorphisms. But such detail is beyond the scope of this post.
Coming back to the point, as previously established, layer III is the genomic analysis sector of the genomic space where many groups are hard at work on the exceedingly difficult problem of elucidating the function of genetic features at the basis of phenotypic traits, including disease. People have been on a formal quest to solve this problem starting two centuries ago (see Gregor Mendel) before it was even understood what DNA was or its sequencing achieved. This pursuit has led to many scientific feats, including the discovery of DNA, its sequencing, as well as its in vivo modification using a bevy of strategies the latest being CRISPR. Yet, the main problem of pinpointing which genetic features are at the basis of disease still stands. So how's the problem being tackled today?
Genetic features identification strategies of layer III
There are many approaches and nuances to consider, which can be classified into three segments: 1) data strategy, 2) identification of causal genetic features, and 3) search and match of known genetic features. The component strategies of these segments are summarized graphically in the figure below along with approximate scores rating technical risk, overall capacity for meaningful actionability and the number of ventures pursuing the strategy.
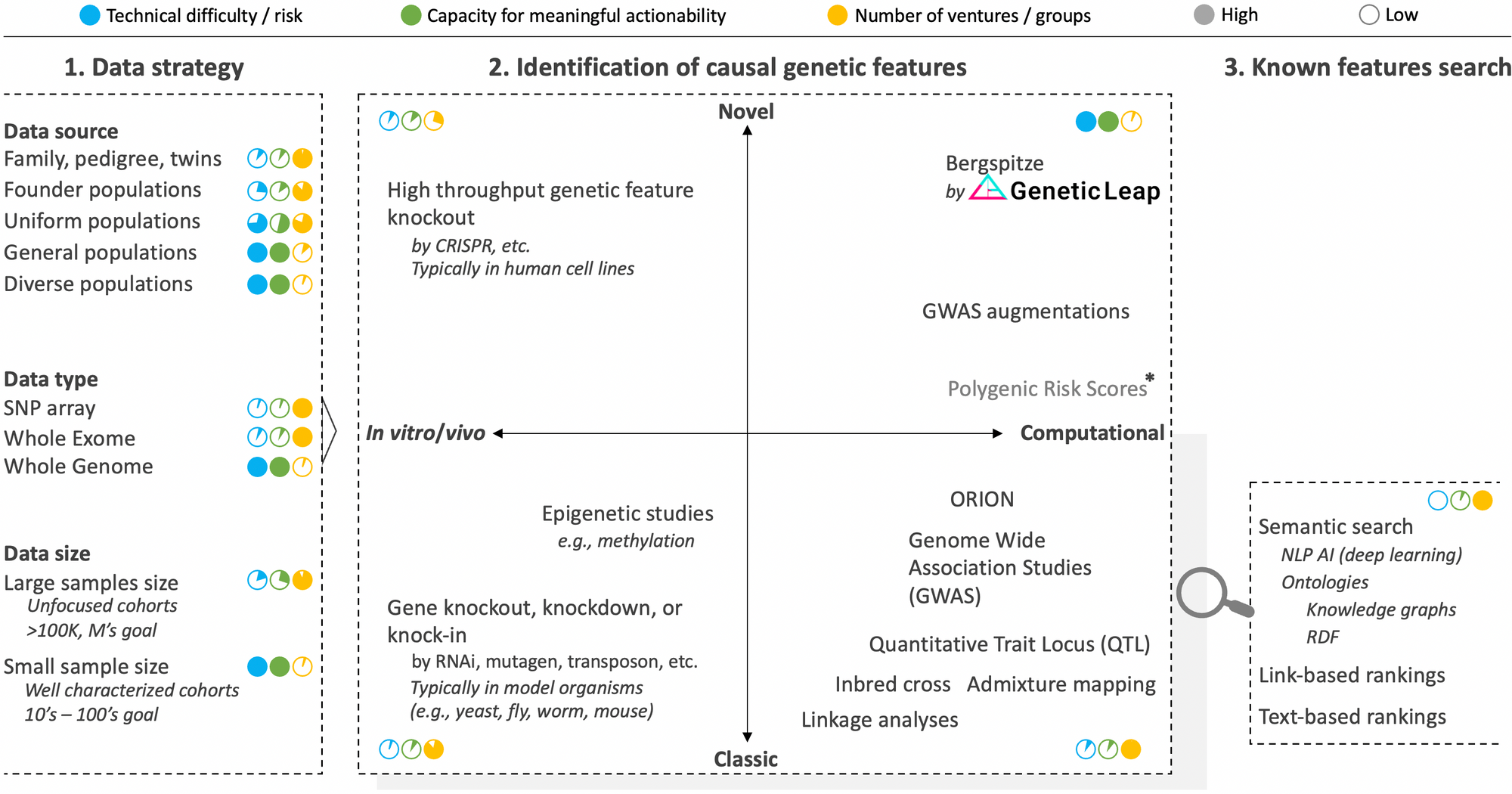
Segment 1 of layer III: Data strategy
The very first decision to make when embarking on the quest to identify the causal genetic features at a basis of a disease phenotype is to decide on the data strategy. As we'll see with the in vivo and in vitro strategies of segment 2, we didn't use to have this luxury. But the advent of powerful cloning and sequencing technologies have increasingly afforded us better design options.
We addressed the questions of which data type to use in a previous post and that of data size in another post still. Respectively, the takeaways were that while the vast majority of ventures currently focus on SNP arrays or whole exomes, it is an unavoidable necessity to analyze the whole genome in its entirety. Likewise, while various players are obsessed with accumulating ever larger sample sizes to achieve statistical significance, we have developed technology to uncover actionable causal genetic features from small cohort sizes, which is critical to neutralize confounding factors, solve disease heterogeneity, and access rare diseases.
This leaves us with the question of data source to discuss, which concerns the background of study participants to enroll. The typical strategy here is to focus on groups where a familial link can help reduce noise in the genome (e.g., twins, family pedigree, and populations with founder effects where there's low genetic variation). Put differently, absent actual inbreeding which has been successfully leveraged in model organisms to identify causal genetic features, the pedigree and founder populations strategies take advantage of the higher degree of inbredness found in certain populations. And they have been effective in solving monogenic diseases (i.e., diseases where only one gene causes the disease like hemophilia) and have helped unearth contributing genes such as SOD1 (ALS), LRRK2 (Parkinson's), and BRCA (breast cancer) in complex diseases. The trouble is that the vast majority of diseases are complexingly polygenic (i.e., many genetic features contribute to the condition), and here, rare genetic features found in family, isolated, or founder cohorts often do not translate to the rest of the population who are much more genetically diverse.
It is therefore imperative that genomic analysis technology be able to handle the inherent outbredness of humanity (even founder populations are outbred compared to the level of inbredness achievable in model organisms). This means that, depending on the region of the world, samples from the general population (any ethnicity) should be analyzed so that the genetic features identified are applicable to all. The ability to tackle small samples sizes mentioned earlier comes in handy here to help negate confounding factors due to differential ancestry.
Segment 2 of layer III: Identification of causal genetic features
There are two primary axes to this segment. The first, in vitro/vivo strategies versus purely computational strategies, which denote whether one begins the genetic feature discovery journey by examining changes in the organism's phenotypic function from disruptions introduced to its genome or by sequencing and examining its intact genome. The second axis concerns whether the strategies are classic or novel.
Classic in vitro/vivo strategies to identify genetic features rely on gene knockout/knockdown methods where a position in the genome is disrupted and the organism observed to see which of its phenotype is in turn impeded. This is a time-honored approach that was used to discover many genes. It requires the use of model organisms since it would be unethical (and criminal) to mutate human beings on purpose in the quest to identify genes, not to mention that the reproduction timelines would not be conducive to the aim. Once the operation is effected in a suitable model organism (e.g., bacteria, yeast, mouse, zebra fish, worm), a corresponding version of the identified gene is then sought in human using homology analysis that is based on the concept that many gene sequences are conserved across species.
Together, classic in vitro/vivo strategies were successful in ascribing a function to a great number of genes and helping to establish an early map of biochemical pathways. They have also solved several monogenic diseases (i.e., diseases where a single gene is responsible for the phenotype like hemophilia or cystic fibrosis). The drawbacks of classic in vitro/vivostrategies for genetic features identification are their lack of high throughput and high resource and time consumption. They also cannot solve complex diseases where multiple genes contribute to the condition. Finally, they were traditionally limited to investigations of the exome, the protein coding component of the genome.
Novel in vitro/vivo strategies are higher throughput versions of the in vitro/vivo strategies where recent genome editing technologies such as CRISPR are leveraged to create knockout mutant libraries at greater scale and potentially across the whole genome. Variations of CRISPR such as CRISPRi and CRISPRa allow for control over gene interference and activation respectively. Further, to speed up examination of mutant cells, AI-assisted computer vision analysis strategies can be enlisted to achieve phenotypic screening at scale. While these innovations have breathed a second life into in vitro/vivo strategies and have great utility especially in drug discovery, they are still inherently unable to identify the genetic features at the basis of complex diseases, which constitute the vast majority of diseases. Only computational strategies have the breadth of logic, scale, and perspective to solve these diseases.
Classic computational strategies began to supplant in vitro/vivo approaches with the advent of DNA sequencing technologies. Through computational abstraction, they allow for direct interrogation of human genomes while maintaining the full integrity of the organisms themselves. They also, as a whole and in theory, have no limits to their logic, scale, and perspective potential to tackle any phenotype imaginable, including complex diseases. The right algorithm just has to be created.
Linkage analyses led a flurry of gene discoveries in the early to mid 90's. Inbred cross and Quantitative Trait Locus approaches have also been successfully applied to discover certain genes, however they tend to require inbreeding and crossing of test subjects (which is out of the question in humans leaving admixture analysis as the best parallel). These older strategies were eventually supplanted by genome wide associated studies (GWAS), a strategy that gained popularity with the greater availability of sequencing technology in the late 2000's. As we explained in another post, GWAS remains the tool of choice to identify genetic features from sequencing data even though is has many well documented flaws, the list of which continues to expand. The main reason for its persistence despite its lack of actionability is that it's a simple and intuitively comprehensible concept for which the genomic field has become convinced that it only needs be fed even more genome sequencing data to finally get it to work properly, thus the aim of many ventures to collect millions of genome samples. We of course believe otherwise.
Novel computational strategies aim to render genomic data analysis more actionable by conceiving novel analysis tools to overcome the limitations of classic computational methods. They can draw from advanced artificial intelligence techniques, but, as those who have attempted to indiscriminately apply in genomics AI techniques that were successful in other areas (e.g., finance, image processing) have found, it should never lose focus of the biological context. This is the type of biology-aware, whole genome analysis AI techniques that Genetic Intelligence is successfully pioneering.
It is also worth pointing out that there are efforts to augment GWAS with various techniques, including Random Forest (RF) to prioritize hits. Finally, Polygenic Risk Scores (PGS) analysis is a new approach that attempts to side step the challenge of genomic noise and small effect sizes by simply combining all possible variant positions into an overall score. There are early reports suggesting that PGS scores may have greater predictive power than variants identified using GWAS. However, the bar is low with GWAS, and one of the most glaring drawbacks for PGS scores is that they cannot even suggest candidate positions in the genome for targeting to remedy a problematic phenotype. Still, they could reveal a useful, even if blunt diagnostic in the overall genomic interrogation arsenal.
Segment 3 of layer III: search and match of known genetic features
As we alluded to in our overview of the genomic space, layer III harbors an important nuance that can be difficult to notice given the strong interplay. Picture writers who write new books and librarians who help catalogue previously written books, make it easy to find how they may relate to other books, and help to unearth potentially important but obscure books.
Similarly, on one side, there is a part focused on innovating better analysis of the genome to make new discoveries (as described in segment 2 above). On the other side, there are those who focus on better searching, matching, aggregating, or drawing new insights from the genetic knowledge that have been recorded over the years in various scientific databases (e.g., ClinVar, PMC, GenBank, dbSNP, gnomAD, OMIM, COREMINE, Gene Ontology, etc.). The latter constitute segment 3 of layer III, which is a useful capability to have for every player in genomic analysis as it is useful to quickly and automatically query the knowledge space to know whether the identified genetic feature has been previously identified and for which disease. The technological hill to surmount is low, and semantic search techniques such as natural language processing (NLP) perfected in other fields can be applied here nearly verbatim.
However, it is critically important to appreciate the clear limitations of the methods of this segment even if it's an artificial intelligence technique being claimed. Namely, if the genetic feature has not been solved and reported (recall that the vast majority of conditions are yet to be solved) then segment 3 strategies are fruitless. The same outcome holds if the genetic feature has been reported but the information is not logged into the databases being searched. Further still, the previously logged discovery could be wrong or have conflicting findings as is often the case with GWAS; therefore, replication studies would need to be carried out for validation if not previously carried out.
Further thoughts
In addition to the data strategy considerations listed in segment 1, the quality of the data also matters a great deal. Very low coverage, mis-labeled, and/or incorrectly formatted data are avoidable but significant impediments often encountered in this space.
The discussion on computational techniques to identify causal genetic features is focused on genomic data from which the vast majority of phenotypic expression is derived. There are approaches that focus on gene expression data, epigenetics data, proteomics data and combination approaches that attempt to integrate more than one type of omics data.
For in vitro/vivo validation, gene knockout/knockdown and rescue experiments are limited to animal models or human cell lines. For validation of hits in humans, options include computational analysis of sequencing data from orthogonal datasets, real world evidence, and clinical trials.
There are often computational components to in vitro/vivo strategies. There is also more to these techniques that go beyond the scope of this post. This book is but one of many resources that provide a more comprehensive view of classic in vitro/vivo gene function elucidation strategies.
It's worth pointing out that computational approaches have benefited greatly from the tremendous body of work established by in vitro/vivo approaches to map an early comprehensive view of many genes, their protein products, and other nodes and edges in the associated biochemical networks.